发布时间:2025-11-28 17: 18: 00
在DNASTAR的多个功能模块中,序列拼接是基因组装、质粒构建、引物验证等工作中常用的一步。若拼接逻辑有误或输入数据存在缺陷,往往会出现错配、断裂、丢碱基、重叠不准确等问题,影响后续注释与分析。特别是在重叠区域未对齐或存在多个候选拼接点时,程序可能自动选择最短路径而忽略了更合理的拼接方式。本文将围绕DNASTAR拼接异常的成因、修复方法及重叠区域的检查步骤展开详细说明,帮助用户精准控制序列拼接结果。
一、DNASTAR序列拼接异常的常见原因
拼接异常多源于输入序列的质量问题或操作中未设定合理参数,导致拼接逻辑偏差或比对失败。
1、序列方向设定不一致
若参与拼接的两段序列方向相反(一个为5'→3',另一个为3'→5'),且未开启自动反向选项,程序将无法识别其真实重叠关系,从而拼接失败或错位。
2、重叠区域存在低质量碱基
尤其是从测序结果导入的FASTA或AB1文件,重叠区域若存在N碱基或质量值过低,会导致程序识别困难,自动舍弃拼接点。
3、缺乏足够重叠长度
默认情况下,SeqMan Pro需15bp以上的连续匹配作为有效拼接依据,若重叠区域太短或存在碱基错配,也将被判定为无法拼接。
4、不同物种或异源序列混拼
混合来源序列往往无显著重叠区,尤其是用户误将多基因或交叉物种片段导入同一工程时,会出现“跳跃拼接”或“断层插入”等异常。
5、操作中跳过了拼接预处理
如未使用【Assemble】或【Auto Assemble】指令,或跳过了预拼接校验步骤,程序将无法识别相邻片段间的接头位点,导致拼接缺失。
二、DNASTAR序列拼接异常的修正方法
当发现拼接图中存在异常断点、空白区或错接时,可以通过以下几种方式进行修正与手动优化。
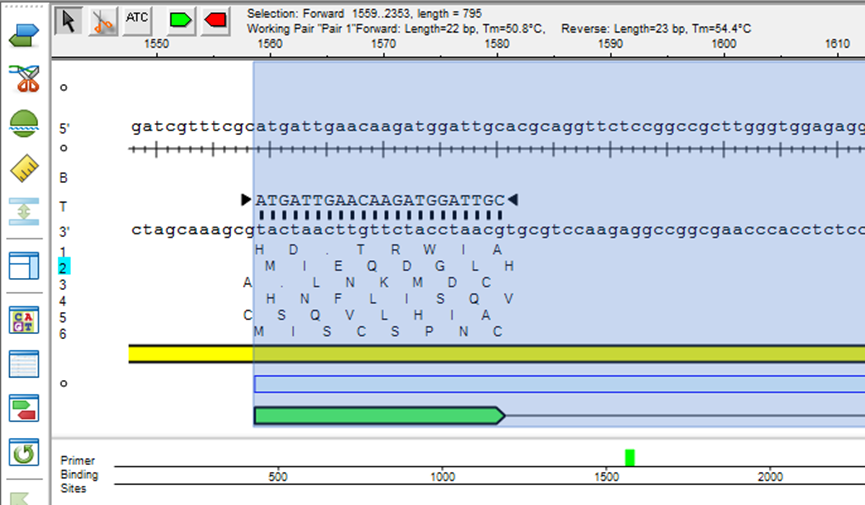
1、使用SeqMan中的Contig Editor检查拼接点
打开项目后切换至【Contig Editor】,在图形视图中选中异常片段,通过颜色标记与错配警告确认是否拼接成功,并定位出重叠错误区域。
2、反转序列方向进行重新装配
右键点击目标片段,选择【Flip Sequence】,或在Import时勾选【Reverse complement】,再重新执行【Assemble】以匹配真实拼接方向。
3、调整拼接敏感度参数
在【Assemble】对话框中,降低“最小匹配长度”与“最大错配比例”,例如设置最小匹配为10bp、允许错配率为10%,可容忍一定变异以实现弱重叠拼接。
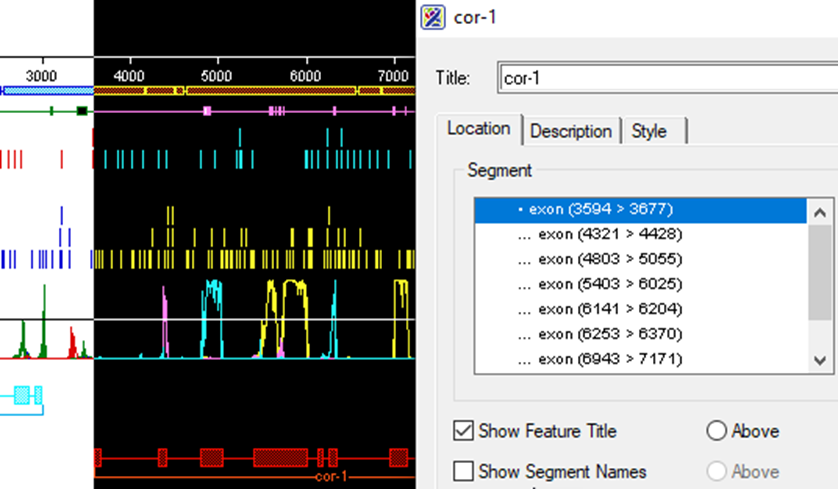
4、手动编辑并插入桥接序列
在确知两个片段之间存在特定链接序列时,可点击【Edit Sequence】插入人工拼接桥段,或通过构建一个合成片段辅助对齐。
5、分批组装后再合并Contig
对于多个短序列拼接失败的情况,可先小范围组装形成多个Contig,再使用【Merge Contigs】功能进行高层次合并。
三、DNASTAR重叠区域的检查方法
正确识别和评估序列之间的重叠区域,是判断拼接是否可靠的关键。以下为具体操作步骤。
1、启用【Alignment View】模式对比重叠段
在Contig中选中目标序列,点击【Alignment】→【Pairwise Alignment】,可查看重叠区的碱基匹配度、错配比例、间隙数量等信息。
2、使用【Dot Plot】可视化检测重叠位置
通过【Tools】→【Dot Plot Viewer】,加载两个待拼接片段,图中对角线即为重叠区,可快速识别反向重叠或错配区域。
3、导出比对表格审阅匹配详情
在【Contig】中选择【Export Alignment Report】,保存为TXT或Excel格式后,可查看每对序列的匹配长度、起止位置、错误率等具体数值。
4、观察拼接点的覆盖深度与质量值
若拼接点覆盖度低于平均水平或出现N、?等低质量碱基标记,表明拼接不稳定,建议人工排查该区域原始测序片段。
5、通过【Sequence Distance Matrix】估算相似度
使用【MegAlign Pro】导入拼接片段并生成相似度矩阵,可辅助判断哪些序列更适合作为拼接对象,避免误接远源序列。
总结
DNASTAR序列拼接的准确性,直接关系到后续注释、比对与功能分析的可信度。面对拼接异常与重叠缺失问题,用户应先排查方向、格式与重叠区质量,再结合图形化工具如Contig Editor与Alignment View进行手动校验与修复。合理设置拼接参数、评估相似度,并在必要时手动插入桥接片段,能显著提升拼接结果的连贯性与科学性。如果你在使用DNASTAR过程中遇到复杂拼接疑难,欢迎留言交流,我们也将持续分享更多高效处理技巧与实战案例。
展开阅读全文
︾
读者也喜欢这些内容:
DNASTAR序列注释怎么编辑 DNASTAR序列注释显示顺序怎么调整
做DNASTAR注释时,很多人前面的问题不是不会加feature,而是改完以后显示越来越乱。名字、范围、方向、翻译这些信息散在不同位置里,如果只在图形视图里盯着一条箭头改,后面很容易漏掉限定词和区段范围。DNASTAR现在更适合处理这件事的入口,主要还是SeqBuilder Pro的Features view。官方说明里把它定义成查看、编辑和管理既有注释的主表格视图,用户既可以编辑文字字段,也可以控制注释是否在Sequence、Linear和Circular这些图形视图里显示。...
阅读全文 >
DNASTAR怎么导入FASTQ数据 DNASTAR导入FASTQ后读长变短怎么排查
做测序数据分析时,FASTQ能不能被顺利导入,以及导入后读长有没有被“莫名变短”,往往决定了后续比对、组装和变异结果是否可信。很多人以为这是文件坏了,其实更常见的是流程选型和剪切参数触发了预处理,或FASTQ质量编码与格式细节不符合软件预期,导致软件把末端当成低质量直接剪掉。把导入路径、配对规则、剪切逻辑三件事一次性核对清楚,后面结果才不会反复返工。...
阅读全文 >
DNASTAR测序分析软件是什么 DNASTAR测序分析能处理哪些数据
很多团队在整理测序结果时,会同时面对三类工作:把原始读段变成可用序列,把序列与参考序列对齐并完成注释,再把结论以可复核的方式交付给课题组或下游分析。DNASTAR常被提到,是因为它以Lasergene为核心,把序列编辑、拼接组装、比对分析与可视化集中在同一套软件体系里,适合用统一口径管理从原始数据到结果文件的全过程。...
阅读全文 >

DNASTAR蛋白结构预测不准确怎么修复 DNASTAR如何调整蛋白结构预测模型
在使用DNASTAR进行蛋白结构预测过程中,常会遇到预测结果不准确、空间折叠异常或活性位点偏移等问题。如果不及时修正,将直接影响下游功能分析、分子对接或疫苗设计等研究任务。因此,深入理解DNASTAR蛋白结构预测不准确怎么修复、DNASTAR如何调整蛋白结构预测模型,对于保证建模质量与科学性尤为关键。...
阅读全文 >


