发布时间:2025-11-28 17: 22: 00
在使用DNASTAR进行蛋白结构预测过程中,常会遇到预测结果不准确、空间折叠异常或活性位点偏移等问题。如果不及时修正,将直接影响下游功能分析、分子对接或疫苗设计等研究任务。因此,深入理解DNASTAR蛋白结构预测不准确怎么修复、DNASTAR如何调整蛋白结构预测模型,对于保证建模质量与科学性尤为关键。

一、DNASTAR蛋白结构预测不准确怎么修复
DNASTAR结构模块依赖模板对比、能量最小化及二级结构辅助预测,因此若模型偏差较大,应从源头分析并按步骤逐项排查。
1、检查输入序列质量
应优先排除测序错误或手动修饰引起的氨基酸错误。可在DNASTAR的序列窗口内使用【Analyze】功能检测不合理残基或缺失信息。
2、合理选择模板结构
使用【Protean 3D】的【Structure Prediction】菜单,通过BLAST筛选高同源度模板,优先选择分辨率小于2.5Å、结构完整、来源物种一致的PDB结构,避免使用残缺或嵌合模板。
3、手动调整比对区域
如自动比对区域发生错位,可点击【Align Sequences】后使用手动调整工具【Adjust Alignment】微调错位的比对片段,保证保守区域准确覆盖。
4、重新优化能量最小化参数
预测完成后,点击【Energy Minimization】,使用AMBER力场并启用“side-chain packing”,调整原子间距离阈值与电荷模型,有助于改善异常扭曲结构。
5、修复活性位点或结构空洞
在模型出现空洞或活性口袋错位时,可手动在【Structure Editor】中插入缺失残基,或使用内置【Refine Structure】进行微调修复。
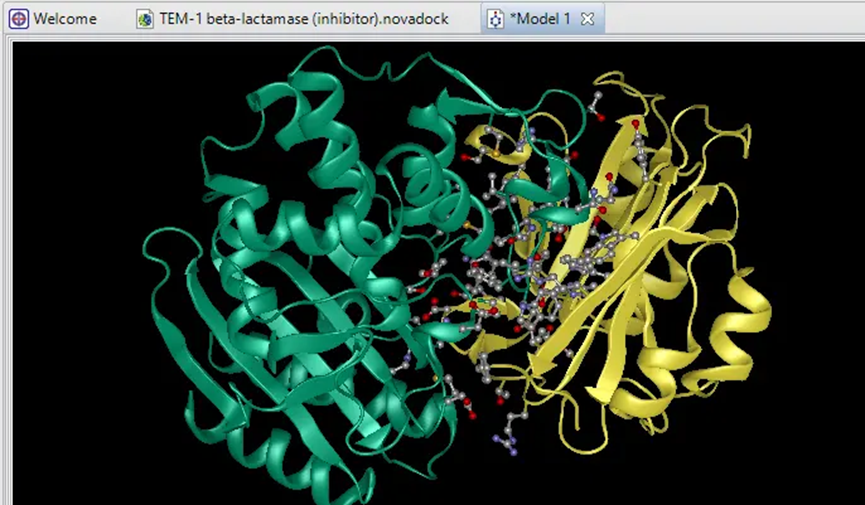
二、DNASTAR如何调整蛋白结构预测模型
调整建模策略不仅能提升结构质量,也有助于提高后续分子对接和功能分析的准确性。
1、启用多算法并行预测
在【Model Building】界面可启用多个算法模块,如SWISS-MODEL模式与Ab Initio模式并行运算,系统将返回多个模型供评估比对。
2、权重调整与评分标准自定义
在【Structure Evaluation】界面内,用户可根据目标应用设定权重偏好,例如提升Ramachandran图打分比重、降低G-factor容差,使筛选模型更贴近研究目标。
3、应用分子动力学模拟校准
通过DNASTAR对接AMBER、GROMACS等MD模块导出模型进行10ns–50ns短时间动力学模拟,分析关键残基稳定性并反馈回模型筛选环节。
4、重新定义结构区域范围
在预测前通过【Domain Definition】划定功能域或折叠模块的起止位置,避免全长建模导致无关区域干扰核心结构成型。
5、结合实验数据微调模型
如拥有CD谱、NMR、质谱交联等实验数据,可通过【Structural Constraints】输入结构约束,辅助提升模型精度。
三、DNASTAR结合分子功能的结构修正思路
在进行蛋白结构预测时,仅仅依靠几何精度往往无法满足功能研究的要求。尤其在配体结合、酶催化或抗原设计等应用中,还需从分子功能出发对结构模型进行二次修正,提升其生物学解释力。
1、围绕活性中心优化残基构象
可通过【Active Site Tools】标记关键位点周围5Å范围内残基,检查其取向、氢键网络与疏水核心是否合理,如存在非自然扭曲,可手动旋转侧链或重新打包侧链链团。
2、模拟底物或配体结合状态
利用【Dock Ligand】工具导入已知配体结构,模拟其结合模式,评估蛋白模型是否存在空间位阻或口袋塌陷,并据此修正α螺旋或β折叠结构框架。
3、关注构象柔性区域
某些蛋白功能依赖结构动态变化,应重点检查连接环、柔性肽段的预测结果是否异常拉伸或折叠,可借助分子动力学对其进行局部预折叠模拟。
4、引导结构向天然构象靠近
使用PDB中同类蛋白复合物结构作为结构约束,对目标模型添加【Positional Restraints】强制收敛关键片段走向,提升整体生物相似度。
5、模拟突变后功能影响
结合结构预测功能与【Mutagenesis】模块,模拟氨基酸突变对结构与口袋构型的影响,用于辅助筛选更稳定或更具活性的变体设计。
这一阶段的结构修正不再仅仅追求几何最优,而是将“是否支持预期功能”作为最终判据,有助于使DNASTAR预测结果从结构模型真正转化为功能研究的起点。
总结
在蛋白建模过程中遇到DNASTAR蛋白结构预测不准确怎么修复、DNASTAR如何调整蛋白结构预测模型的问题时,应遵循逐步排查、手动校正与实验结合的原则。通过精准筛选模板、细化对齐策略、启用多重预测机制,并结合分子动力学与结构评价手段,可有效提升模型的可靠性,为后续生物信息分析与实验研究提供坚实支撑。
展开阅读全文
︾
读者也喜欢这些内容:
DNASTAR Contig拼接结果靠谱吗 DNASTAR Contig覆盖度太低怎么改
做Sanger序列拼接时,你会反复遇到两件事:第一是DNASTAR Contig拼接结果靠谱吗,第二是DNASTAR Contig覆盖度太低怎么改。前者本质是证据链够不够,包含覆盖深度、冲突分布、读段质量与一致性;后者多半与前处理裁剪、拼接阈值、读段方向与数据量有关。把查看口径与重拼步骤固定下来,结果往往更可解释、更好复现。...
阅读全文 >
DNASTAR序列拼接出现异常怎么修正 DNASTAR序列拼接重叠区域如何检查
在DNASTAR的多个功能模块中,序列拼接是基因组装、质粒构建、引物验证等工作中常用的一步。若拼接逻辑有误或输入数据存在缺陷,往往会出现错配、断裂、丢碱基、重叠不准确等问题,影响后续注释与分析。特别是在重叠区域未对齐或存在多个候选拼接点时,程序可能自动选择最短路径而忽略了更合理的拼接方式。本文将围绕DNASTAR拼接异常的成因、修复方法及重叠区域的检查步骤展开详细说明,帮助用户精准控制序列拼接结果。...
阅读全文 >

DNASTAR怎么看测序质量的准确性 DNASTAR怎么拆分测序图重叠峰
在测序结果的分析过程中,准确性与可视化解读是两大核心关注点。DNASTAR作为一款专业的生物信息分析软件,不仅支持多种测序数据的处理,还在质量评估和重叠峰识别方面提供了较为全面的功能。在实验流程数字化、自动化的背景下,理解如何判断测序的质量以及有效拆分重叠峰,不仅关乎数据是否可靠,更影响后续分析与报告的精度。...
阅读全文 >


