发布时间:2026-01-27 00: 00: 00
做Sanger序列拼接时,你会反复遇到两件事:第一是DNASTAR Contig拼接结果靠谱吗,第二是DNASTAR Contig覆盖度太低怎么改。前者本质是证据链够不够,包含覆盖深度、冲突分布、读段质量与一致性;后者多半与前处理裁剪、拼接阈值、读段方向与数据量有关。把查看口径与重拼步骤固定下来,结果往往更可解释、更好复现。
一、DNASTAR Contig拼接结果靠谱吗
判断靠谱与否,不要只看是否拼成一条consensus,更要看拼接依据是不是集中在高质量重叠区,冲突是不是能被原始峰图解释。SeqMan Pro在预拼接阶段会做端部质量裁剪、向量与污染筛查,再基于读段重叠进行拼接并生成报告,你需要把这些环节的输出当作判读依据来用。
1、先用覆盖视图把证据密度看清楚
在工程里打开覆盖相关视图或报告,重点看深度在关键区域是否连续、是否存在长段落只有单条读段支撑,以及正反向读段是否都覆盖到目标区,覆盖图和覆盖报告本身就是你判断是否需要补测的第一手依据。
2、把冲突集中区单独拎出来核对而不是平均看
遇到局部错配不要急着改结论,先在Trace Data窗口里打开对应区域,看冲突是否来自低质量尾端、混峰、或某条读段方向反了;把冲突图与原始峰图对齐后,你会更容易判断是测序质量问题还是样本真实变异。
3、确认拼接阈值是否与你的数据质量一致
拼接能否成立取决于重叠区的匹配率与阈值设置,Minimum Match Percentage决定了重叠要达到的匹配比例,阈值设得偏高会让读段更难并入同一contig,阈值设得偏低又会放大误拼风险,判断靠谱时要把当时的阈值设置一并纳入复盘。
4、检查预拼接裁剪是否把有效信息剪掉
端部裁剪与向量筛查会提升整体质量,但也可能在读段本来就短或峰图衰减快的情况下,把本可用于重叠的区域剪掉;如果你发现读段之间只差一点点就能形成重叠,可以先回到裁剪参数,把过于激进的裁剪放宽后再重新拼一次再评估。
5、用重新拼接而不是手工拖拽来验证可重复性
当你怀疑存在错位或假连接时,优先在Project Summary里选中目标contig后执行【Contig】→【Reassemble Contig】,让软件只在该contig内部重新排列与对齐读段;如果多次重拼都稳定得到相同布局,通常比一次手工调整更能说明结果可靠。
二、DNASTAR Contig覆盖度太低怎么改
覆盖度低不等于软件算错,更多时候是数据量不足、方向不全、或参数把读段排除在拼接之外。SeqMan Pro的重拼命令不会自动把Unassembled Sequences里的新数据加进来,所以要提升覆盖度,思路要分成两步,先让更多读段能进入候选集合,再让它们能通过阈值并入contig。
1、先确认是不是读段没有被纳入拼接而不是拼不上
打开Unassembled Sequences窗口看是否还有未组装读段,如果有,先按正常组装流程加入并触发组装,而不是只对现有contig反复重拼;同时注意【Sequence】菜单里的【Add One】会绕过端部裁剪与向量扫描等预处理,容易把低质量读段带入或把应当处理的步骤跳过,导致覆盖表现更乱。
2、把端部裁剪参数调到能保留有效重叠区的水平
进入【Project】→【Parameters】后找到End Trimming相关设置,先把目标定清楚,宁可保留能形成重叠的中等质量区,也不要只留下很短的高质量片段导致读段之间没有足够重叠;调整后重新组装并对比覆盖图变化,确保改动确实带来覆盖提升。
3、按重叠长度与匹配率两条线同步调阈值
当覆盖低是因为读段并不差但始终无法并入contig,优先回到拼接阈值,检查Minimum Match Percentage是否过高导致轻微噪声就被拒绝,同时核对你对重叠长度的预期是否合理;每次只改一项并重新组装,避免同时改多项导致你无法判断是哪一项在起作用。
4、用延伸contig端部恢复被剪掉的末端数据
如果覆盖缺口集中在contig两端,先尝试【Contig】→【Extend Contig Ends】恢复端部被裁剪的数据,再回看覆盖图是否把边缘缺口补上;这一步适合解决边缘覆盖不足和端部无法对齐的问题,但恢复后要同步检查冲突是否随之增加。
5、覆盖缺口在中间时优先补测或补方向而不是强行拼
当缺口出现在中段且缺口两侧读段质量正常,通常说明你缺少跨越该区域的读段,最直接的做法是补一条能跨过缺口的引物步移读段,或补齐反向读段;如果你为了“拼成一条”去降低阈值或强行合并contig,往往会换来更难解释的冲突与后续验证成本。
6、覆盖报表与对齐视图深度不一致时先统一口径再决策
有时你会看到Coverage Report里的Depth与Alignment View里肉眼数到的读段数不一致,这种差异需要先按软件说明理解其统计口径,再决定是否真的需要补测或重拼,避免因为报表口径差异误判为覆盖不足。
三、DNASTAR Contig冲突区怎么核对
覆盖与靠谱最终都会落到冲突区的解释上,冲突区处理得好,你的consensus就更可用。SeqMan Pro提供了冲突图、峰图查看、冲突拆分提示与手工连接等能力,你要做的是先用证据决定是否应拆分,再决定是否要合并,而不是反过来。
1、先用Conflicts Graph找出冲突是否呈现规律
在Trace Data窗口里显示冲突相关图形,观察冲突是零散分布还是在某一段形成连续模式;连续冲突更像是错位、混样或重复序列引发的布局问题,零散冲突更像是个别低质量碱基或真实变异。
2、对连续冲突先尝试系统提示的拆分线索
在Project Summary里选中contig后可用【Contig】→【Suggest Conflict Splits】定位可能需要拆分的位置,拆分与重拼的目的不是让图更好看,而是让每个contig内部的读段一致性更高,覆盖与冲突都更容易解释。
3、需要合并contig时先对齐重叠再评估是否值得Join
对两个可能相邻的contig,优先用【Contig】菜单的对齐与连接相关命令让软件计算重叠得分,再看是否达到Minimum Match Percentage阈值;只有当重叠区的对齐质量足够且冲突能被峰图解释时,合并才更稳。
4、把低质量尾端造成的伪冲突先处理掉再做结论
如果冲突集中在某些读段尾端,先回到裁剪参数或对单条读段做端部处理,让明显不可靠的尾端不再参与共识计算,再看冲突是否自然消失;这一步能避免你在不可靠数据上过度解读。
总结
DNASTAR Contig拼接结果靠谱吗,DNASTAR Contig覆盖度太低怎么改,判断与改动都建议围绕同一套证据链推进:先看覆盖图与覆盖报告,再看冲突图与峰图能否解释差异,必要时用【Contig】→【Reassemble Contig】验证布局可重复性;覆盖低时先把读段纳入与预处理口径理顺,再按阈值与裁剪逐项调整,缺口跨不过去就补测补方向。这样你得到的不是一条看起来连起来的序列,而是一条你能说明它为什么可信的共识序列。
展开阅读全文
︾
读者也喜欢这些内容:
DNASTAR注释文件怎么导入 DNASTAR注释显示不全怎么解决
DNASTAR注释文件怎么导入,DNASTAR注释显示不全怎么解决,遇到的多数不是同一个毛病:有的文件本身没有注释,有的是注释导入了但显示轨道被折叠或隐藏,还有的是版本问题导致轨道区域空白。处理时先把注释从哪里来讲清楚,再按软件里真实的显示逻辑逐项核对,基本都能定位到具体一步。...
阅读全文 >
DNASTAR授权文件在哪里导入 DNASTAR授权校验失败怎么处理
DNASTAR授权文件在哪里导入,DNASTAR授权校验失败怎么处理这类问题,多数不是文件放错目录,而是授权入口选错了位置或授权类型填错了信息。Lasergene这套产品常见的授权方式是通过DNASTAR Navigator里的License Manager完成授权录入,独立授权填产品密钥,网络授权填许可服务器的主机名或IP地址,离线场景再走手动授权流程,把网页生成的Keys粘贴回授权对话框即可。...
阅读全文 >
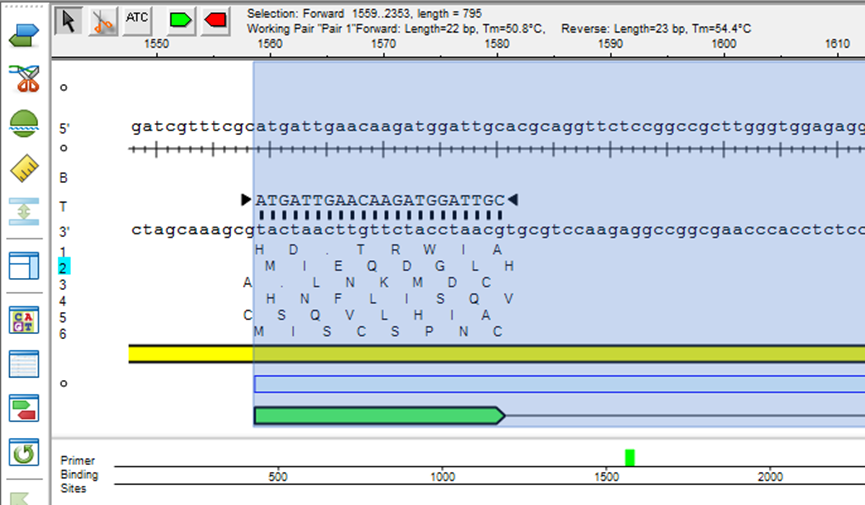
DNASTAR序列拼接出现异常怎么修正 DNASTAR序列拼接重叠区域如何检查
在DNASTAR的多个功能模块中,序列拼接是基因组装、质粒构建、引物验证等工作中常用的一步。若拼接逻辑有误或输入数据存在缺陷,往往会出现错配、断裂、丢碱基、重叠不准确等问题,影响后续注释与分析。特别是在重叠区域未对齐或存在多个候选拼接点时,程序可能自动选择最短路径而忽略了更合理的拼接方式。本文将围绕DNASTAR拼接异常的成因、修复方法及重叠区域的检查步骤展开详细说明,帮助用户精准控制序列拼接结果。...
阅读全文 >


