发布时间:2026-01-28 00: 00: 00
做分子序列分析时,参考序列选得不合适,后面再怎么调参数也容易出现对不齐、缺口乱跳、变异位点看起来不可信等问题。围绕“DNASTAR参考序列怎么选,DNASTAR参考比对错位怎么纠正”,更实用的做法是先把参考口径定稳,再用软件里现成的对齐、修剪、方向校正与局部重排功能,把错位收回到可解释范围内。
一、DNASTAR参考序列怎么选
参考序列不是越长越好,而是要能代表你这批数据的目标区域与版本口径。选之前先想清楚你是做多序列比对与变异对照,还是做参考引导的组装与SNP分析,两类任务对参考的要求不一样。
1、先区分你用的是MegAlign Pro的参考,还是SeqMan NGen的模板参考
在MegAlign Pro里,参考序列可以是任意长度与类型的序列,包括DNA、RNA、蛋白,并且更多用于对齐锚定、变异对照与特征映射;而在SeqMan NGen或SeqMan Ultra里,参考更像模板,用来引导短读段或片段拼装到contig上,两者概念不同,别混着用。
2、做多序列比对时,优先选“版本清晰、注释完整、覆盖你关注区段”的那条
如果你要把实验序列与公开数据库序列对齐后看变异,建议优先使用来源明确的注释参考,这样后续做变异比对或把参考特征映射到其它序列时,解释成本更低。MegAlign Pro也明确把参考用于variant analysis与features mapping这两类典型工作流。
3、做参考引导组装时,模板要与测序对象一致,避免跨菌株或跨亚型硬套
模板与真实样本差异过大,会表现为覆盖断裂、错配集中、插缺口异常增多,后面在SeqMan Pro里看变异也会偏离真实情况。SeqMan NGen的LoadTemplate说明也强调模板会作为SeqMan Pro里显示与分析的reference基础。
4、优先处理好参考的方向与注释,再把它作为参考导入
如果参考序列本身需要补注释,SeqMan NGen文档给出的思路是先在SeqBuilder里打开并完成注释,再作为模板输入到组装流程,这样后续在SeqMan Pro里下钻时更顺手。
5、在MegAlign Pro里把参考设为“对齐锚点”要按算法来选入口
如果你准备用MAFFT并让参考参与对齐引导,可以先选中目标序列,再走【Sequences】→【Use as Reference】,随后用【Align】→【Align Using MAFFT】启动;也可以直接用【Align】→【Align with Options】,在Using里选MAFFT并在Reference Sequence里指定参考,然后点Align。
二、DNASTAR参考比对错位怎么纠正
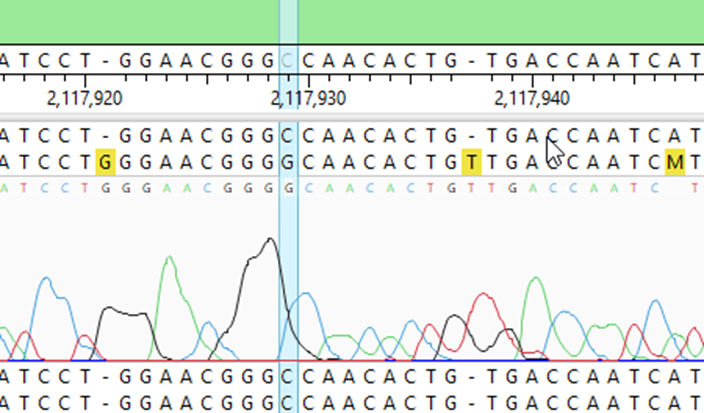
比对错位通常不是单一原因,常见是方向反了、端部未修剪、局部插缺口放错位置、参考不适配或对齐算法与数据类型不匹配。处理时建议先用“可逆操作”把全局形态拉回正常,再做局部精修。
1、先检查序列方向,必要时做反向互补
当你看到大段区域整体对不上、错配密集但又呈规律性,优先怀疑方向问题。在MegAlign Pro里可以选中序列后用工具栏的反向互补功能,或在序列上右键选择反向互补,再重新跑对齐。
2、把端部噪声剪掉再对齐,先解决“起点不齐”
Sanger序列或拼接后的片段两端常有低质量或引物残留,容易把对齐算法拖偏。先做端部修剪,再重新对齐,通常比一上来手动塞缺口更省事。MegAlign Pro提供单条序列修剪入口,例如通过【Sequences】→【Trim Sequence】或工具栏修剪按钮进入修剪对话框。
3、全局已经乱了就先取消对齐,回到未对齐状态重跑
当你发现缺口已经被手动改得很碎,或不同算法之间来回切换后整体结构不可读,建议直接用【Align】→【Unalign All】把序列恢复到未对齐状态,再从头用同一套算法与参数重跑,这一步是把混乱“清零”的快捷方式。
4、只是一小段错位,用SeqMan Pro的局部重排功能更稳
如果你是在SeqMan Pro的Alignment View里看到某个小区域缺口放偏或出现GC压缩导致的小范围错位,可以框选共识序列的目标区段后走【Edit】→【Adjust Alignment】做局部重排,这个功能专门用来优化小区域对齐,并且可以用【Edit】→【Undo】回退;需要手工对齐时可以用工具栏的Banana工具。
5、在MegAlign Pro里需要“对着参考看错位”时,先把参考指定正确再算变异
错位有时是你把参考换了但仍沿用旧的变异视角,表现为差异点位置看着不对。MegAlign Pro允许切换参考,并说明基于旧参考计算过的variants不会丢失;如果你想改为对共识算变异,可以用【Sequences】→【Stop Using as Reference】移除参考关系再计算。
6、还在错位就回到参考本身,确认参考版本与目标区段没有选错
如果你选的是全基因组参考但你的数据只覆盖某个片段,或你用的是不同构建版本的参考,算法会在大量缺失区硬拉缺口,视觉上就像整体错位。此时更务实的是换成同区段、同版本、同方向的参考,再重复前面“修剪→设参考→重跑对齐”的顺序。
三、DNASTAR比对结果如何复核
把错位拉回正常后,还需要用一套固定检查动作确认结果能用,否则很容易把算法产物当成真实变异或真实同源关系。复核做得扎实,后面出树、做注释映射或统计变异才不容易返工。
1、固定一个基准视图先看全局一致性,再下钻局部
在MegAlign Pro里先用Overview或Sequences view看缺口是否集中在端部或低复杂区,再决定是重跑对齐还是只做局部修补,避免在全局没稳定前就去抠单个位点。
2、用参考特征映射校验坐标是否合理
如果参考带注释特征,设为参考后可以把特征映射到其它序列,再反向检查关键基因或功能区是否落在预期位置,这比只看碱基差异更容易发现“对齐漂移”。
3、变异对照时明确你比较的是参考还是共识
同一批数据用参考对照与用共识对照,变异列表的坐标与解释口径会不同。需要改口径时,在MegAlign Pro里按流程移除或更换参考,再重新计算对应的variants,避免把两套口径混在同一张表里。
4、把关键区域导出成报告或截图留痕,方便复盘与交付
当你要把比对结论交给同事或写进实验记录,建议把关键视图导出并记录参考来源与对齐算法名,这样后续出现争议时能快速定位是数据、参考还是参数导致的差异。
总结
围绕“DNASTAR参考序列怎么选,DNASTAR参考比对错位怎么纠正”,先把参考的类型与用途分清,再按方向校正、端部修剪、取消重跑与局部重排的顺序处理,通常能把错位收敛到可解释范围。最后用参考特征映射与变异口径复核一遍,能显著降低后续出树、做注释或报差异时的返工概率。
展开阅读全文
︾
读者也喜欢这些内容:
DNASTAR Contig拼接结果靠谱吗 DNASTAR Contig覆盖度太低怎么改
做Sanger序列拼接时,你会反复遇到两件事:第一是DNASTAR Contig拼接结果靠谱吗,第二是DNASTAR Contig覆盖度太低怎么改。前者本质是证据链够不够,包含覆盖深度、冲突分布、读段质量与一致性;后者多半与前处理裁剪、拼接阈值、读段方向与数据量有关。把查看口径与重拼步骤固定下来,结果往往更可解释、更好复现。...
阅读全文 >
DNASTAR更新到新版本要注意什么 DNASTAR更新后旧工程打不开怎么办
做DNASTAR升级时,真正麻烦的往往不是安装包能不能装上,而是升级后发现旧工程打不开,或者能打开但资源丢失、注释不全、路径断链。想把风险压住,做法是先把版本与数据口径固定,再按应用类型把工程文件与可交换格式同时备份,最后用一台干净环境验证打开与导出链路是否通畅。...
阅读全文 >

DNASTAR转录组数据分析结果异常怎么办 DNASTAR如何校准基因表达量计算
在RNA测序数据分析中,基因表达量的准确计算与解读直接影响后续的差异分析、功能富集以及调控网络构建。DNASTAR Lasergene Genomics Suite作为一款整合了序列比对、表达定量和可视化功能的分析平台,广泛用于转录组数据的建库质控、比对分析和表达挖掘。然而,在实际使用过程中,用户常会遇到表达量异常、结果偏差大、样本间差异不合理等问题。本文将围绕“DNASTAR转录组数据分析结果异常怎么办”以及“DNASTAR如何校准基因表达量计算”两个核心问题进行系统说明。...
阅读全文 >
DNASTAR软件闪退是什么原因 DNASTAR运行时提示内存不足如何解决
DNASTAR作为一款常用于生物序列分析、比对、进化树构建和结构预测的综合平台,其图形化操作、数据密集度和计算强度都较高,部分用户在运行过程中可能会遇到闪退、内存不足、程序无响应等情况。本文将围绕DNASTAR闪退的常见原因进行分析,并提供实用的应对方法,特别针对“内存不足”问题,提出系统级与软件级的解决思路,帮助用户更顺畅地进行生物信息分析工作。...
阅读全文 >


