发布时间:2025-11-28 17: 16: 00
在基因组学与分子生物学研究中,序列比对是极为基础且高频的操作,尤其在分析同源性、构建系统树、探测保守区域、设计引物等场景中起到关键作用。DNASTAR作为一款集成化的生物信息分析软件,其核心模块MegAlign与SeqBuilder等功能均支持多种序列比对算法。为了提高分析效率并避免误操作,用户需掌握DNASTAR序列比对的具体流程、算法适用范围以及软件本身的性能上限。
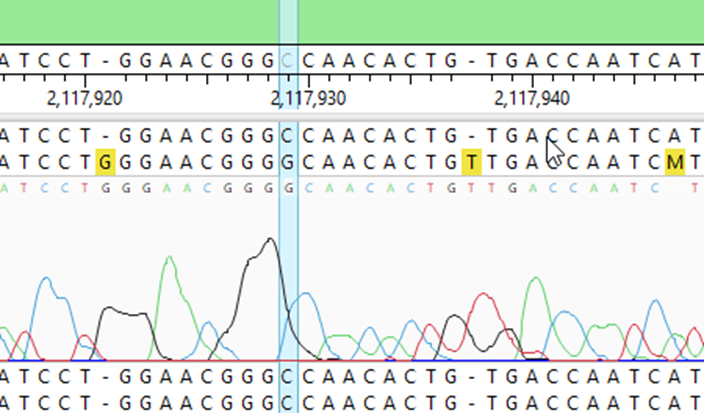
一、DNASTAR怎么比对序列
DNASTAR软件中的MegAlign模块支持多种经典比对算法,包括Clustal W、Clustal Omega、MAFFT、MUSCLE、Jotun Hein等,覆盖了从快速比对到高保真比对的需求。具体比对操作流程如下:
1、导入待比对序列
打开MegAlign Pro,点击菜单栏【File】→【New Project】,选择【Add Sequences】添加FASTA、GenBank、.seq、.gb等格式的核酸或蛋白质序列文件,可批量导入。
2、设置比对算法
在顶部工具栏选择【Alignment Algorithm】,用户可根据需求选择Clustal Omega(适合大规模序列)、MAFFT(兼顾速度与准确度)或MUSCLE(适合进化树分析)。
3、配置参数选项
在【Alignment Settings】中可设定Gap penalty、Substitution Matrix(如BLOSUM62、DNAfull)、迭代次数、树构建方式等,确保比对策略与目标分析一致。
4、执行比对并查看结果
点击【Align】按钮启动比对过程,完成后在主视图中可观察多序列比对结果图、保守性评分、碱基一致性热图等,并可切换至【Phylogenetic Tree】选项卡生成系统发育树。
5、导出比对数据
可通过【File】→【Export Alignment】导出为FASTA、Nexus、Phylip、Clustal格式的比对结果,用于下游分析软件或文献插图。
二、DNASTAR最多能比对多少条序列
在实际应用中,DNASTAR的序列比对能力不仅受限于软件设计本身,还受到硬件性能与内存限制的影响。
1、MegAlign标准版支持数量
传统MegAlign模块在32位系统下理论支持序列上限为200条以内,每条长度建议不超过5000个碱基;在64位系统和高性能工作站下,最多可稳定比对约500条中等长度序列。
2、MegAlign Pro扩展能力更强
MegAlign Pro模块采用64位架构优化,并支持GPU加速和多线程处理,实际可比对序列数量上限在1000条左右,单条长度可超过100kb。若仅比对短序列(如引物或保守区段),可扩展至1500条以上。
3、算法对上限影响
Clustal W对序列数量敏感,适合200条以内;Clustal Omega与MAFFT具备并行计算能力,适合500条以上大规模比对;Jotun Hein因计算复杂度较高,适用于不超过100条的深度进化分析。
4、内存和显卡配置的作用
在运行MegAlign Pro时,建议系统配置至少为16GB内存、四核处理器,并支持OpenGL 2.0以上的独立显卡,以保证比对速度与可视化响应稳定。
5、超大数据比对替代方案
若比对序列规模超出DNASTAR限制,可使用DNASTAR与CLC、Geneious等软件联动,或将部分数据分批比对后再合并对齐结果;也可转向基于命令行的MAFFT或Clustal Omega独立工具完成预处理。
三、DNASTAR序列比对技巧与注意事项
为了提高比对质量与操作效率,建议用户在日常分析中遵循以下实践:
1、进行类型分组比对
避免将蛋白质与核酸序列混合比对,MegAlign会默认识别类型,混合文件可能导致比对失败或字符错位。
2、保持输入序列方向一致
在SeqBuilder中预处理序列时应统一正负链方向,防止反向互补影响比对准确性。
3、定期保存比对工程文件
比对任务运行时间较长时,建议中途保存【.msa】或【.megaproj】工程文件,防止意外断电或软件崩溃造成数据丢失。
4、选用合适的保守性评分标准
可在【View】→【Conservation Score】中切换至Shannon entropy、Consensus percentage等指标,帮助快速识别突变热点或保守区域。
5、利用颜色编码增强可读性
MegAlign支持按碱基、氨基酸理化性质上色显示,可通过【Color Scheme】选项启用Heatmap、Identity、Hydrophobicity等风格,提升比对解读效率。

总结
DNASTAR在序列比对功能上提供了灵活算法、可视化操作与良好的可拓展性,适合日常科研中对小到中等规模的核酸与蛋白序列进行精准比对。在掌握基本操作流程的基础上,合理选择比对算法、评估输入数据量与硬件配置,能更充分释放软件性能,帮助用户更高效完成多序列分析、保守性判断与系统树构建等工作。
展开阅读全文
︾
读者也喜欢这些内容:
DNASTAR Contig拼接结果靠谱吗 DNASTAR Contig覆盖度太低怎么改
做Sanger序列拼接时,你会反复遇到两件事:第一是DNASTAR Contig拼接结果靠谱吗,第二是DNASTAR Contig覆盖度太低怎么改。前者本质是证据链够不够,包含覆盖深度、冲突分布、读段质量与一致性;后者多半与前处理裁剪、拼接阈值、读段方向与数据量有关。把查看口径与重拼步骤固定下来,结果往往更可解释、更好复现。...
阅读全文 >
DNASTAR安装教程怎么操作 DNASTAR安装后启动报错怎么解决
DNASTAR装完打不开、启动就报错,十有八九不是安装包坏了,而是许可模式、网络路径、旧版本残留这几件事没对齐。更稳妥的做法是把安装与授权当成同一条流程来走:先确认你用的是单机许可还是网络许可,再按对应路径装客户端与许可组件,最后用License Manager做一次可见的授权验证,能把大多数问题提前挡在第一次启动之前。...
阅读全文 >
DNASTAR是什么软件 dnastar软件有哪些功能和特点
在生物信息学日益发展的今天,科研人员面对的是海量的DNA、RNA、蛋白质等序列数据,如何高效、准确地进行比对、注释、变异分析和结构预测,成为科研进程中的关键一环。DNASTAR软件正是为此应运而生的一款综合性分子生物学分析平台,它集成了多个功能模块,覆盖从原始数据处理到高级可视化的各个分析阶段,为基因组学、转录组学、蛋白组学等领域提供了高效、稳定的技术支撑。...
阅读全文 >
DNASTAR如何生成三维模型 DNASTAR蛋白三维结构显示异常怎么办
在结构生物学和分子建模分析中,蛋白质的三维模型是理解其功能机制、药物结合位点及突变影响的关键依据。作为经典的分子生物信息学平台,DNASTAR套件中的Protean 3D模块提供了直接从序列生成、预测并可视化蛋白三维结构的功能。实际操作中,部分用户也会遇到模型异常或显示不全的问题,本文将详细解析DNASTAR如何生成三维模型,以及在三维结构显示异常时应如何排查与修复。...
阅读全文 >


