发布时间:2025-11-28 17: 02: 00
在进行分子生物学研究时,常常需要比对多条DNA、RNA或蛋白质序列,寻找保守区域、突变位点或结构变异。DNASTAR作为一款集成性极强的序列分析平台,提供了多种比对工具和参数调控方式,适用于多序列比对、局部比对、结构比对等多种场景。在实际使用中,不仅需要掌握其比对入口和操作流程,还要明确如何针对结果中出现的空缺进行解释与调整,以获得更可靠的分析结论。
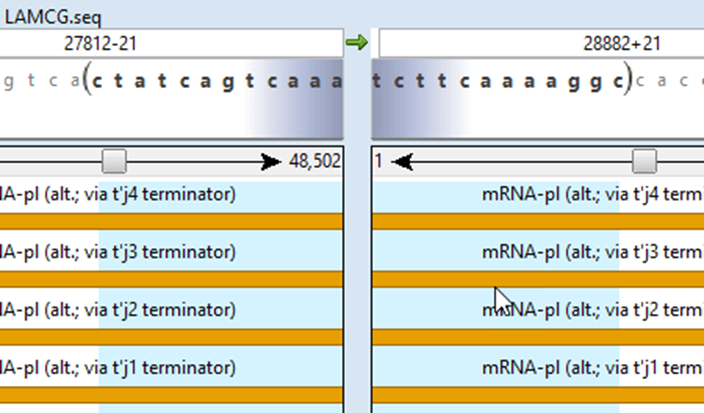
一、DNASTAR如何比对多条序列
多序列比对旨在同时对比三条及以上的核酸或蛋白序列,从而识别它们之间的保守性与变异性
1、导入序列集合
在SeqMan Pro中点击【File】选择【New Project】,随后通过【Add Sequences】导入需要比对的多条序列文件,支持FASTA、GenBank、.seq等多种格式
2、启动多序列比对模块
在项目窗口中点击【Align】按钮,自动跳转至MegAlign模块。选择比对算法如ClustalW、Muscle或MAFFT,并加载所有目标序列
3、设置比对参数
根据物种亲缘关系、序列长度、缺失比例等调整Gap Penalty、Substitution Matrix等参数,确保对齐的灵敏性与准确性
4、运行并可视化比对
点击【Align】启动比对流程,系统将生成比对图谱与差异统计表,用户可通过色彩区分突变位置、缺口区域及一致性片段
5、保存比对结果
在菜单栏选择【Export Alignment】,可导出对齐结果为FASTA格式或图像形式,用于后续结构注释与功能分析
二、DNASTAR序列比对结果有空缺怎么办
在比对结果中出现空缺(gap)是由于插入/缺失(indel)事件引起的,这种情况需结合生物学背景及分析目标具体处理
1、分析空缺位置的功能意义
可在GeneQuest中查看对应位置是否处于编码区、保守基序、结构域内,若影响功能区域,应重点关注并进一步验证
2、调整Gap设置优化对齐
回到MegAlign中重新设定Gap Open Penalty与Gap Extension Penalty,例如降低Gap惩罚值有助于保留真实缺口而非人为排除
3、使用结构比对交叉验证
若为蛋白序列,可使用Protean 3D模块加载PDB结构进行三维比对,辅助判断缺口是否为构象差异引起
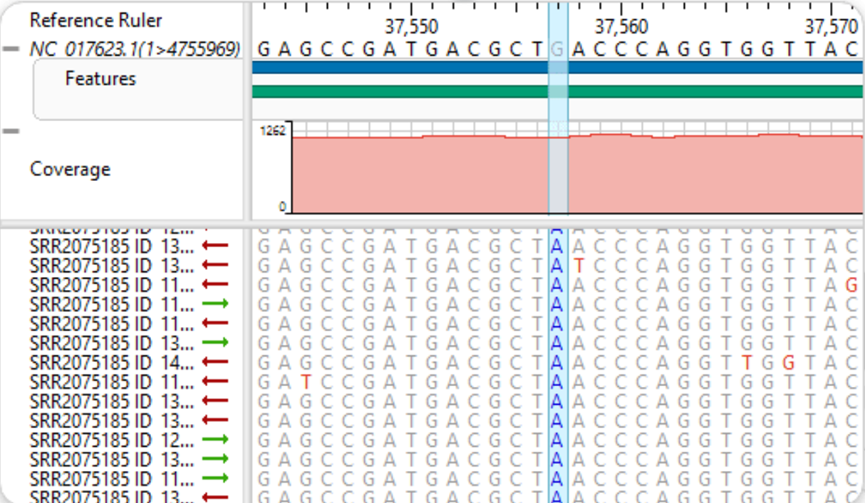
4、手动优化特定区域对齐
对个别比对质量较差的片段,可启用手动编辑模式,在Alignment视图中拖动残基对齐位置,微调关键位点一致性
5、输出空缺位点统计
在比对结果窗口点击【Report】生成空缺统计表,可帮助用户一键识别空缺频繁区域并分析其生物学相关性
三、DNASTAR比对与空缺分析的实用组合技巧
多序列比对与空缺判读往往是联合使用的两个步骤,不同研究目的下的设置方式亦有所不同
1、针对病毒变异分析
选择MAFFT算法增强短序列比对稳定性,空缺位置建议保留并作为候选突变位点分析,适合COVID-19、流感等快速演化物种研究
2、用于物种进化树构建
建议使用ClustalW配合Neighbor-Joining算法,空缺区域不宜过度处理,以避免影响系统发育信号识别
3、在蛋白家族比对中
优先选择BLOSUM62矩阵处理保守性评分,空缺多发生在环区或无序区,应结合结构图共同评估保守性
4、配合批量处理
在Lasergene核心平台中使用脚本批量运行多个数据集的比对任务,同时输出空缺统计报告,提升分析效率
5、优化结果导出格式
对于存在大量空缺的结果,应使用Nexus或Phylip格式导出,便于下游建树或模型训练工具识别与兼容
总结
在使用DNASTAR进行多条序列比对时,用户应充分理解每个比对算法的适用场景,合理设置参数,才能准确捕捉保守区域与变异特征。当比对结果出现空缺时,不能简单忽略或删除,而应结合生物学背景进行解读与优化。通过掌握比对工具的细节功能和空缺处理技巧,可以显著提高序列分析的科学性与可重复性,为基因功能注释、结构预测或进化研究奠定坚实基础。
展开阅读全文
︾
读者也喜欢这些内容:
DNASTAR测序峰图怎么校对 DNASTAR测序峰图杂峰怎么处理
做Sanger数据时,很多人最头疼的不是装配跑不起来,而是峰图看着像能读,真到校对时又总觉得不踏实。前面一段峰形还算整齐,到了后面开始叠峰、虚峰、拖尾,或者某个位置明明像有变异,又拿不准到底是样本真的混了,还是读段质量已经掉下去了。DNASTAR的SeqMan Ultra本来就把这类工作放在装配后的分析阶段来做,官方资料也明确提到,它支持查看ABI色谱图、检查冲突、手动修剪末端,并对装配结果做碱基层级和contig层级的编辑。想把峰图校对顺,关键不是一上来就改碱基,而是先把峰图、质量、比对位置和杂峰类型分开判断。...
阅读全文 >
DNASTAR授权文件在哪里导入 DNASTAR授权校验失败怎么处理
DNASTAR授权文件在哪里导入,DNASTAR授权校验失败怎么处理这类问题,多数不是文件放错目录,而是授权入口选错了位置或授权类型填错了信息。Lasergene这套产品常见的授权方式是通过DNASTAR Navigator里的License Manager完成授权录入,独立授权填产品密钥,网络授权填许可服务器的主机名或IP地址,离线场景再走手动授权流程,把网页生成的Keys粘贴回授权对话框即可。...
阅读全文 >

DNASTAR转录组数据分析结果异常怎么办 DNASTAR如何校准基因表达量计算
在RNA测序数据分析中,基因表达量的准确计算与解读直接影响后续的差异分析、功能富集以及调控网络构建。DNASTAR Lasergene Genomics Suite作为一款整合了序列比对、表达定量和可视化功能的分析平台,广泛用于转录组数据的建库质控、比对分析和表达挖掘。然而,在实际使用过程中,用户常会遇到表达量异常、结果偏差大、样本间差异不合理等问题。本文将围绕“DNASTAR转录组数据分析结果异常怎么办”以及“DNASTAR如何校准基因表达量计算”两个核心问题进行系统说明。...
阅读全文 >

DNASTAR怎么看测序质量的准确性 DNASTAR怎么拆分测序图重叠峰
在测序结果的分析过程中,准确性与可视化解读是两大核心关注点。DNASTAR作为一款专业的生物信息分析软件,不仅支持多种测序数据的处理,还在质量评估和重叠峰识别方面提供了较为全面的功能。在实验流程数字化、自动化的背景下,理解如何判断测序的质量以及有效拆分重叠峰,不仅关乎数据是否可靠,更影响后续分析与报告的精度。...
阅读全文 >


